
ALIMTA 500 mg, poudre pour solution à diluer pour perfusion, boîte de 1 flacon de poudre de 500 mg
Retiré du marché le : 25/07/2025
Dernière révision : 30/09/2020
Taux de TVA : 2.1%
Laboratoire exploitant : LILLY FRANCE
Source :

Mésothéliome pleural malin
ALIMTA, en association avec le cisplatine, est indiqué dans le
traitement des patients atteints de mésothéliome pleural malin non
résécable et qui n'ont pas reçu de chimiothérapie antérieure.
Cancer bronchique non à petites cellules
ALIMTA, en association avec le cisplatine, est indiqué dans le
traitement en première ligne des patients atteints de cancer bronchique
non à petites cellules localement avancé ou métastatique, dès lors que
l'histologie n'est pas à prédominance épidermoïde (voir rubrique Propriétés pharmacodynamiques).
ALIMTA est indiqué en monothérapie dans le traitement de maintenance du cancer bronchique non à petites cellules, localement avancé ou métastatique immédiatement à la suite d'une chimiothérapie à base de sel de platine, dès lors que l'histologie n'est pas à prédominance épidermoïde chez les patients dont la maladie n'a pas progressé (voir rubrique Propriétés pharmacodynamiques).
ALIMTA est indiqué en monothérapie dans le traitement en seconde ligne des patients atteints de cancer bronchique non à petites cellules, localement avancé ou métastatique, dès lors que l'histologie n'est pas à prédominance épidermoïde (voir rubrique Propriétés pharmacodynamiques).
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Composition.
Allaitement (voir rubrique Grossesse et allaitement).
Association concomitante avec le vaccin contre la fièvre jaune (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Le pémétrexed peut entraîner une dépression médullaire, qui se manifeste par une neutropénie, une thrombopénie et une anémie (ou une pancytopénie) (voir rubrique Effets indésirables). La myélosuppression est généralement un effet toxique dose-limitant. Les patients doivent être surveillés pour myélosuppression pendant le traitement, et le pémétrexed ne doit pas être administré aux patients tant que leur taux de polynucléaires neutrophiles (PNN) n'est pas revenu à une valeur≥ 1 500 cellules/mm 3 et leur taux de plaquettes à une valeur ≥ 100 000 cellules/mm3. Les réductions de doses pour les cycles ultérieurs dépendent du taux de PNN et de plaquettes au nadir et de la toxicité non hématologique maximale observés lors du cycle précédent (voir rubrique Posologie et mode d'administration).
Une moindre toxicité et une réduction des toxicités hématologiques et non-hématologiques de grade 3/4 telles que neutropénie, neutropénie fébrile et infections avec neutropénies de grade 3/4 ont été rapportées lorsqu'une prémédication par acide folique et vitamine B12 était administrée. Tous les patients traités par le pémétrexed doivent donc être informés de la nécessité de prendre de l'acide folique et de la vitamine B12 comme mesure prophylactique afin de réduire la toxicité liée au traitement (voir rubrique Posologie et mode d'administration).
Des réactions cutanées ont été rapportées chez des patients n'ayant pas reçu de corticothérapie préalable. Une prémédication par dexaméthasone (ou équivalent) peut réduire l'incidence et la sévérité des réactions cutanées (voir rubrique Posologie et mode d'administration).
Un nombre insuffisant de patients présentant une clairance de la créatinine < 45 mL/min a été étudié. Par conséquent, l'utilisation du pémétrexed chez les patients présentant une clairance de la créatinine inférieure à 45 mL/min n'est pas recommandée (voir rubrique Posologie et mode d'administration).
Les patients atteints d'une insuffisance rénale faible à modérée (clairance de la créatinine comprise entre 45 mL/min et 79 mL/min) doivent éviter de prendre des anti-inflammatoires non stéroïdiens (AINS) tel que l'ibuprofène et l'acide acétylsalicylique (> 1,3 g par jour) les deux jours avant, le jour même et les deux jours suivant l'administration de pémétrexed (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Chez les patients atteints d'insuffisance rénale légère à modérée susceptibles de recevoir un traitement par le pémétrexed, les AINS à demi-vie longue doivent être interrompus pendant au moins cinq jours avant, le jour même, et au moins les deux jours suivant l'administration de pémétrexed (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Des événements rénaux graves, y compris une insuffisance rénale aiguë, ont été rapportés avec le pémétrexed en monothérapie ou en association avec d'autres agents cytotoxiques. La plupart des patients chez lesquels ces événements sont survenus présentaient des facteurs de risque rénaux, incluant une déshydratation, une hypertension ou un diabète préexistants. Des cas de diabète insipide néphrogénique et de nécrose tubulaire rénale ont également été rapportés après commercialisation en cas d'utilisation du pémétrexed en monothérapie ou en association avec d'autres agents cytotoxiques. La plupart de ces événements ont disparu après l'arrêt du pémétrexed. Les patients doivent être régulièrement surveillés pour détecter une nécrose tubulaire aiguë, une diminution de la fonction rénale ainsi que les signes et symptômes du diabète insipide néphrogénique (dont l'hypernatrémie, par exemple).
L'effet d'un troisième secteur liquidien, tel qu'un épanchement pleural ou une ascite, sur le pémétrexed n'est pas entièrement défini. Une étude de phase 2 du pémétrexed conduite chez 31 patients atteints de tumeurs solides et ayant un troisième secteur liquidien stable a démontré qu'il n'y avait pas de différence en termes de concentrations plasmatiques normalisées et de clairance du pémétrexed, comparés aux patients n'ayant pas de troisième secteur liquidien. Ainsi, une ponction évacuatrice d'une collection du troisième secteur liquidien avant l'administration de pémétrexed devrait être envisagée, mais peut ne pas être nécessaire.
En raison de la toxicité gastro-intestinale du pémétrexed administré en association avec le cisplatine, une déshydratation sévère a été observée. En conséquence, les patients doivent recevoir un traitement anti-émétique adéquat et une hydratation appropriée, avant et/ou après l'administration du traitement.
Des effets cardiovasculaires graves, y compris infarctus du myocarde, et des effets cérébrovasculaires ont été peu fréquemment rapportés pendant les études cliniques avec le pémétrexed, habituellement lorsque celui-ci est administré en association avec un autre agent cytotoxique. La plupart des patients chez lesquels ces évènements ont été observés avaient des facteurs de risque cardiovasculaire préexistants (voir rubrique Effets indésirables).
L'immunodépression est fréquente chez les patients cancéreux. En conséquence, l'utilisation concomitante de vaccins vivants atténués n'est pas recommandée (voir rubriques Contre-indications et Interactions avec d'autres médicaments et autres formes d'interactions).
Le pémétrexed peut entraîner des anomalies du matériel génétique. Il doit être conseillé aux hommes de ne pas concevoir d'enfant durant leur traitement et dans les 6 mois qui suivent son arrêt. Des mesures contraceptives ou l'abstinence sont recommandées. Une conservation de sperme peut être conseillée aux hommes avant de débuter le traitement en raison du risque de stérilité irréversible.
Les femmes en âge de procréer doivent utiliser un moyen de contraception efficace pendant le traitement par pémétrexed (voir rubrique Grossesse et allaitement).
Des cas de pneumopathie radique ont été rapportés chez des patients traités par radiothérapie, soit avant, pendant ou après une chimiothérapie par pémétrexed. Une attention particulière devra être portée à ces patients et il conviendra d'agir avec précaution lors de l'utilisation d'autres agents radiosensibilisants.
Des cas de réactivation de zone antérieurement irradiée ont été rapportés chez des patients préalablement traités par radiothérapie des semaines ou des années auparavant.
Excipients
ALIMTA 100 mg poudre pour solution à diluer pour perfusion
Ce médicament contient moins de 1 mmol de sodium par flacon (23 mg), c'est-à-dire qu'il est essentiellement « sans sodium ».
ALIMTA 500 mg poudre pour solution à diluer pour perfusion
Ce médicament contient 54 mg de sodium par flacon, ce qui équivaut à 2,7 % de l'apport alimentaire quotidien maximal recommandé par l'OMS de 2 g de sodium par adulte.
Résumé du profil de sécurité
Les effets indésirables les plus fréquemment rapportés chez les patients traités par pémétrexed en monothérapie ou en association, ont été une dépression médullaire à type d'anémie, neutropénie, leucopénie, thrombopénie ; ainsi que des toxicités gastro-intestinales, à type d'anorexie, nausées, vomissements, diarrhées, constipation, pharyngite, mucite et stomatite. D'autres effets indésirables incluent : toxicités rénales, élévation des aminotransférases, alopécie, fatigue, déshydratation, éruption cutanée, infection/sepsis et neuropathie. Des effets rarement observés incluent : syndrome de Stevens- Johnson et nécrolyse épidermique toxique.
Liste tabulée des effets indésirables
Le tableau 4 présente les événements indésirables, quel que soit le lien de causalité, associés au pémétrexed utilisé soit en monothérapie soit en association avec le cisplatine, issus des études pivotales d'enregistrement (JMCH, JMEI, JMBD, JMEN et PARAMOUNT) et de la notification spontanée post-commercialisation.
Les événements indésirables sont listés par classe de système d'organes selon la classification MedDRA. La convention suivante a été utilisée pour la classification par fréquence : très fréquent :≥ 1/10 ; fréquent : ≥ 1/100, < 1/10 ; peu fréquent : ≥ 1/1 000, < 1/100 ; rare : ≥ 1/10 000, < 1/1 000 ; très rare : < 1/10 000 et fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Tableau 4. Fréquences des événements indésirables de tout grade, quel que soit le lien de causalité, issus des études pivotales d'enregistrement : JMEI (ALIMTA versus docétaxel), JMDB (ALIMTA et cisplatine versus GEMZAR et cisplatine), JMCH (ALIMTA plus cisplatine versus cisplatine), JMEN et PARAMOUNT (pémétrexed plus le meilleur traitement symptomatique versus placebo plus le meilleur traitement symptomatique) et de la notification spontanée post-commercialisation.
| Classe de système d'organes (MedDRA) | Très fréquent | Fréquent | Peu fréquent | Rare | Très rare | Fréquence indéter- minée |
| Infections et infestations | Infectiona Pharyngite | Sepsisb | | | Dermo- hypoder- mite | |
| Affections hématologiques et du système lymphatique | Neutropénie Leucopénie Diminution de l'hémoglobine | Neutropénie fébrile Diminution du nombre de plaquettes | Pancytopénie | Anémie hémolytique auto-immune | | |
| Affections du système immunitaire | | Hypersensi- bilité | | Choc anaphylactique | | |
| Troubles du métabolisme et de la nutrition | | Déshydrata- tion | | | | |
| Affections du système nerveux | Trouble du goût Neuropathie périphérique motrice Neuropathie périphérique sensorielle Sensations vertigineuses | Accident vasculaire cérébral Accident vasculaire cérébral ischémique Hémorragie intracrânienne | ||||
| Affections oculaires | Conjonctivite Sécheresse oculaire Hypersécré- tion lacry- male Kératocon- jonctivite sèche Œdème palpébral Maladie de la surface oculaire | |||||
| Affections cardiaques | Insuffisance cardiaque Arythmie | Angor Infarctus du myocarde Coronaropa- thie Arythmie su- praventricu- laire | ||||
| Affections vasculaires | Ischémie périphériquec | |||||
| Affections respiratoires, thoraciques et médiastinales | Embolie pulmonaire Pneumopathie interstitiellebd | |||||
| Affections gastro- intestinales | Stomatite Anorexie Vomissement Diarrhée Nausées | Dyspepsie Constipation Douleur abdominale | Hémorragie rectale Hémorragie gastro- intestinale Perforation intestinale Œsophagite Colitee | |||
| Affections hépatobiliaires | Elévation de l'alanine aminotrans- férase Elévation de l'aspartate aminotrans- férase | Hépatite | ||||
| Affections de la peau et du tissus sous-cutané | Eruption cuta- née Exfoliation cutanée | Hyperpig- mentation Prurit Erythème polymorphe Alopécie Urticaire | Erythème | Syndrome de Stevens- Johnsonb Nécrolyse épider- mique toxiqueb Pem- phigoïde Dermatite bulleuse Epider- molyse bulleuse acquise Œdème érythéma- teuxf Pseudocel- lulite Dermatite Eczéma Prurigo | ||
| Affections du rein et des voies urinaires | Dimunition de la clairance de la créatinine Augmentation de la créatininémiee | Insuffisance rénale Diminution du débit de filtration glomérulaire | Diabète insipide néphrogé- nique Nécrose tubulaire rénale | |||
| Troubles généraux et anomalies au site d'administration | Fatigue | Pyrexie Douleur Œdème Douleur thoracique Inflammation des muqueuses | ||||
| Investigations | Elévation de la gamma- glutamyl- transférase | |||||
| Lésions, intoxications et complications liées aux procédures | Œsophagite radique Pneumopathie radique | Phénomène de rappel de la zone irradiée |
b avec des cas d'issue fatale
c conduisant parfois à une nécrose des extrémités
d avec une insuffisance respiratoire
e observé uniquement en association avec le cisplatine
f principalement des membres inférieurs
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration - voir Annexe V.
SURVEILLANCE du traitement :
Avant chaque perfusion :
- NFS, plaquettes.
- Fonction hépatique.
- Fonction rénale.
PENDANT LE TRAITEMENT, les patients doivent être régulièrement surveillés pour détecter une nécrose tubulaire aiguë, une diminution de la fonction rénale ainsi que les signes et symptômes du diabète insipide néphrogénique (dont l'hypernatrémie, par exemple).
PRESCRIRE en association au traitement :
- de l'acide folique (350 à 1000 µg) : 5 doses dans les 7 jours qui précèdent la 1ère injection, 1 fois par jour pendant le traitement, 1 fois par jour pendant les 21 jours après la dernière injection.
- une injection IM de vitamine B12 (1000 µg) dans la semaine précédant la première injection, puis environ toutes les 9 semaines.
Femmes en âge de procréer / Contraception chez les hommes et les femmes
Les femmes en âge de procréer doivent utiliser un moyen de contraception efficace pendant le traitement par pémétrexed.
Le pémétrexed peut entraîner des anomalies du matériel génétique. Il doit être conseillé aux hommes de ne pas concevoir d'enfant durant leur traitement et dans les 6 mois qui suivent son arrêt. Des mesures contraceptives ou l'abstinence sont recommandées.
Grossesse
Il n'y a pas de données concernant l'utilisation du pémétrexed chez la femme enceinte, cependant le pémétrexed, comme d'autres anti-métabolites, est suspecté d'entraîner des malformations lorsqu'il est administré pendant la grossesse. En effet, les études chez l'animal mettent en évidence une toxicité de la reproduction (voir rubrique Données de sécurité précliniques). Le pémétrexed ne doit pas être utilisé chez la femme enceinte, sauf cas de nécessité absolue, après avoir évalué le bénéfice pour la mère et le risque pour le fœtus (voir rubrique Mises en garde et précautions d'emploi).
Allaitement
On ne sait pas si le pémétrexed est excrété dans le lait maternel et un risque d'effets indésirables chez le nouveau-né allaité ne peut être exclu. L'allaitement doit être interrompu au cours du traitement avec le pémétrexed (voir rubrique Contre-indications).
Fertilité
Une conservation de sperme peut être conseillée aux hommes avant de débuter le traitement en raison du risque de stérilité irréversible.
Le pémétrexed est principalement éliminé sous forme inchangée dans les urines par sécrétion tubulaire et dans une moindre mesure par filtration glomérulaire. L'administration concomitante de médicaments néphrotoxiques (par exemple : les aminosides, les diurétiques de l'anse, les dérivés du platine, la ciclosporine) peut potentiellement diminuer la clairance du pémétrexed. Ces associations doivent être utilisées avec prudence. Si nécessaire, la clairance de la créatinine sera étroitement surveillée.
L'administration concomitante de substances également secrétées au niveau tubulaire (par exemple : le probénécide, la pénicilline) pourrait diminuer la clairance du pémétrexed. Des précautions doivent être prises lorsque ces médicaments sont associés au pémétrexed. Si nécessaire, la clairance de la créatinine devra être étroitement surveillée.
Chez les patients ayant une fonction rénale normale (clairance de la créatinine ≥ 80 mL/min), de fortes doses d'anti-inflammatoires non stéroïdiens (AINS, tel que l'ibuprofène > 1 600 mg/jour) et d'acide acétylsalicylique à dose plus élevée (≥ 1,3 g par jour) peuvent diminuer l'élimination du pémétrexed et, par conséquent, augmenter la survenue d'effets indésirables dus au pémétrexed. Des précautions doivent donc être prises lors de l'administration concomitante de fortes doses d'AINS ou d'acide acétylsalicylique et de pémétrexed chez les patients ayant une fonction rénale normale (clairance de la créatinine ≥ 80 mL/min).
Chez les patients atteints d'une insuffisance rénale faible à modérée (clairance de la créatinine comprise entre 45 mL/min et 79 mL/min), l'administration concomitante de pémétrexed et d'AINS (par exemple l'ibuprofène) ou d'acide acétylsalicylique à dose plus élevée doit être évitée les deux jours avant, le jour même et les deux jours suivant l'administration de pémétrexed (voir rubrique Mises en garde et précautions d'emploi).
En l'absence de données concernant les éventuelles interactions avec les AINS à demi-vie longue, tels que le piroxicam ou le rofecoxib, leur administration concomitante avec le pémétrexed chez les patients ayant une insuffisance rénale faible à modérée doit être interrompue pendant au moins cinq jours avant, le jour même, et au moins les deux jours suivant l'administration de pémétrexed (voir rubrique Mises en garde et précautions d'emploi). Si l'administration concomitante d'AINS est nécessaire, les patients doivent faire l'objet d'une surveillance étroite de la toxicité, en particulier gastrointestinale et de la myélosuppression du pémétrexed.
Le pémétrexed fait l'objet d'un métabolisme hépatique limité. Les résultats d'études in vitro sur microsomes hépatiques humains suggèrent que le pémétrexed n'inhiberait pas de manière cliniquement significative la clairance métabolique des médicaments métabolisés par les iso-enzymes CYP3A, CYP2D6, CYP2C9 et CYP1A2.
Interactions communes à tous les cytotoxiques
En raison de l'augmentation du risque thrombotique chez les patients cancéreux, le recours à un traitement anticoagulant est fréquent. La grande variabilité intra-individuelle de la coagulabilité au cours des maladies, à laquelle s'ajoute l'éventualité d'une interaction entre les anticoagulants oraux et les cytotoxiques, imposent s'il est décidé de traiter le patient par anticoagulants oraux, d'augmenter la fréquence des contrôles de l'INR (International Normalised Ratio).
Association concomitante contre-indiquée : vaccin contre la fièvre jaune : risque de maladie vaccinale généralisée mortelle (voir rubrique Contre-indications).
Association concomitante déconseillée : vaccins vivants atténués (excepté le vaccin contre la fièvre jaune, pour lequel l'association concomitante est contre-indiquée) : risque de maladie vaccinale généralisée éventuellement mortelle. Ce risque est majoré chez les sujets déjà immunodéprimés par leur maladie sous-jacente. Utiliser un vaccin inactivé lorsqu'il existe (poliomyélite) (voirrubrique Mises en garde et précautions d'emploi).
Posologie
ALIMTA doit être administré uniquement sous le contrôle d'un médecin qualifié dans l'utilisation des chimiothérapies anticancéreuses.
ALIMTA en association avec le cisplatine
La posologie recommandée d'ALIMTA est de 500 mg/m2
de surface corporelle, en perfusion intraveineuse de 10 minutes, le
premier jour de chaque cycle de 21 jours. La posologie recommandée de
cisplatine est de 75 mg/m2 de
surface corporelle en perfusion de 2 heures, débutée environ 30 minutes
après la fin de la perfusion de pémétrexed, le premier jour de chaque
cycle de 21 jours.
Les patients doivent recevoir un traitement anti-émétique adéquat et une hydratation appropriée, avantet/ou après la perfusion de cisplatine (se référer au Résumé des Caractéristiques du Produit du cisplatine pour des recommandations posologiques spécifiques).
ALIMTA en monothérapie
Chez les patients traités pour cancer bronchique non à petites cellules
et qui ont reçu une chimiothérapie antérieure, la posologie recommandée
d'ALIMTA est de 500 mg/m2 de surface corporelle, en perfusion intraveineuse de 10 minutes, le premier jour de chaque cycle de 21 jours.
Prémédication
Afin de réduire la survenue et la sévérité des réactions cutanées, une
corticothérapie devra être administrée la veille, le jour même et le
lendemain de l'administration du pémétrexed. La posologie doit être équivalente à 4 mg de dexaméthasone par voie orale, deux fois par jour (voir rubrique Mises en garde et précautions d'emploi).
Afin de réduire la toxicité du pémétrexed, les patients traités doivent recevoir également une supplémentation en vitamines (voir rubrique Mises en garde et précautions d'emploi). Les patients doivent prendre par voie orale de l'acide folique ou une association polyvitaminique contenant de l'acide folique (350 à 1 000 microgrammes) quotidiennement. Au moins cinq doses d'acide folique doivent être prises dans les 7 jours qui précèdent la première injection de pémétrexed, et les patients doivent continuer cette supplémentation pendant toute la durée du traitement et pendant 21 jours après la dernière injection de pémétrexed. Les patients doivent également recevoir une injection intramusculaire de vitamine B12 (1 000 microgrammes) dans la semaine précédant la première dose de pémétrexed puis une fois tous les trois cycles. Les injections ultérieures de vitamine B12 peuvent avoir lieu le même jour que l'administration de pémétrexed.
Surveillance
Avant chaque administration de pémétrexed, une numération formule
sanguine complète (NFS) avec mesure du taux de plaquettes doit être
réalisée. Un bilan biochimique sera réalisé avant chaque administration
de la chimiothérapie pour évaluer les fonctions hépatique et rénale.
Avant le début de chaque cycle de chimiothérapie, le nombre absolu de polynucléaires neutrophiles (PNN) doit être supérieur ou égal à 1 500 cellules/mm3 et le nombre de plaquettes supérieur ou égal à 100 000 cellules/mm3.
La clairance de la créatinine doit être supérieure ou égale à 45 mL/min.
Le taux de bilirubine totale doit être inférieur ou égal à 1,5 fois la limite supérieure de la normale. Les taux de phosphatases alcalines (PA), d'aspartate aminotransférase (ASAT ou SGOT) et d'alanine aminotransférase (ALAT ou SGPT) doivent être inférieurs ou égaux à 3 fois la limite supérieure de la normale. Des taux de phosphatases alcalines, d'ASAT et d'ALAT inférieurs ou égaux à 5 fois la limite supérieure de la normale sont acceptables en cas de métastases hépatiques.
Ajustements de la dose
Au début d'un nouveau cycle, les ajustements de dose s'appuieront sur
la numération formule sanguine au nadir et la toxicité non
hématologique maximale observée au cours du cycle précédent. Le
traitement peut être différé le temps nécessaire à la récupération. Dès
récupération, les patients doivent être à nouveau traités selon les
recommandations des tableaux 1, 2 et 3, qui concernent ALIMTA utilisé
en monothérapie et en association avec le cisplatine.
| Tableau 1 - Tableau de modification des doses d'ALIMTA (en monothérapie ou en association) et de cisplatine - Toxicités hématologiques | |
| Au nadir : PNN < 500 /mm3 et plaquettes ≥ 50 000 /mm3 | 75 % de la dose précédente (pour ALIMTA et le cisplatine) |
| Au nadir : plaquettes < 50 000 /mm3 quel que soit le taux de PNN | 75 % de la dose précédente (pour ALIMTA et le cisplatine) |
| Au nadir : plaquettes < 50 000 /mm3 avec saignementa, quel que soit le taux de PNN | 50 % de la dose précédente (pour ALIMTA et le cisplatine) |
a Ces critères répondent à la définition des saignements ≥ Grade 2 selon les Critères Communs de Toxicité (CTC) du National Cancer Institute (v2.0; NCI 1998)
En cas de toxicités non-hématologiques ≥ grade 3 (à l'exclusion d'une neurotoxicité), le traitement par ALIMTA doit être suspendu jusqu'à résolution à un niveau inférieur ou égal au niveau initial du patient avant traitement. Le traitement doit être poursuivi selon les recommandations du tableau 2.
| Tableau 2 - Tableau de modification des doses d'ALIMTA (en monothérapie ou en association) et de cisplatine - Toxicités non-hématologiques a,b | ||
| | Dose d'ALIMTA (mg/m²) | Dose de cisplatine (mg/m²) |
| Toute toxicité de grade 3 ou 4, excepté mucite | 75 % de la dose précédente | 75 % de la dose précédente |
| Toute diarrhée nécessitant une hospitalisation (quel que soit le grade) ou diarrhée de grade 3 ou 4 | 75 % de la dose précédente | 75 % de la dose précédente |
| mucite de grade 3 ou 4 | 50 % de la dose précédente | 100 % de la dose précédente |
a Critères Communs de Toxicité (CTC) du National Cancer Institute (v2.0; NCI 1998)
b A l'exclusion d'une neurotoxicité
En cas de neurotoxicité, il est recommandé d'ajuster les doses d'ALIMTA et de cisplatine comme précisé dans le tableau 3. Les patients doivent arrêter le traitement si une neurotoxicité de grade 3 ou 4 est observée.
| Tableau 3 - Tableau de modification des doses d'ALIMTA (en monothérapie ou en association) et de cisplatine - Neurotoxicité | ||
| Grade CTC a | Dose d'ALIMTA (mg/m²) | Dose de cisplatine (mg/m²) |
| 0 - 1 | 100 % de la dose précédente | 100 % de la dose précédente |
| 2 | 100 % de la dose précédente | 50 % de la dose précédente |
a Critères Communs de Toxicité (CTC) du National Cancer Institute (v2.0; NCI 1998)
Le traitement par ALIMTA doit être arrêté si le patient présente une toxicité hématologique ou non- hématologique de grade 3 ou 4 après 2 réductions de dose ou immédiatement si une neurotoxicité de grade 3 ou 4 est observée.
Populations particulières
Sujets âgés
Au cours des essais cliniques, il n'a pas été mis en évidence de risque
plus élevé d'effets indésirables chez les patients de 65 ans et plus
comparativement aux patients de moins de 65 ans. Des réductions de
doses autres que celles recommandées pour l'ensemble des patients ne
sont pas nécessaires.
Population pédiatrique
Il n'existe pas d'utilisation justifiée d'ALIMTA dans la population
pédiatrique dans le mésothéliome pleural malin et le cancer bronchique
non à petites cellules.
Insuffisants rénaux (formule standard de Cockcroft et Gault ou taux de filtration glomérulaire mesuré par la méthode de clairance plasmatique Tc99m-DTPA)
Le pémétrexed est essentiellement éliminé sous forme inchangée dans les urines. Dans les études cliniques, des ajustements de doses autres que celles préconisées pour l'ensemble des patients n'ont pas été nécessaires chez les patients dont la clairance de la créatinine était ≥ 45 mL/min. Chez les patients ayant une clairance de la créatinine < 45 mL/min, les données sont insuffisantes ; l'utilisation du pémétrexed n'est donc pas recommandée chez ces patients (voir rubrique Mises en garde et précautions d'emploi).
Insuffisants hépatiques
Aucune relation entre le taux d'ASAT (SGOT), d'ALAT (SGPT) ou de
bilirubine totale et la pharmacocinétique du pémétrexed n'a été
identifiée. Toutefois, il n'a pas été conduit d'étude spécifique chez
des patients ayant une atteinte hépatique avec un taux de bilirubine
supérieur à 1,5 fois la limite supérieure de la normale et/ou un taux
d'aminotransférases supérieur à 3 fois la limite supérieure de la
normale (en l'absence de métastases hépatiques) ou supérieur à 5 fois
la limite supérieure de la normale (en cas de métastases hépatiques).
Mode d'administration
ALIMTA doit être administré par voie intraveineuse. ALIMTA doit être administré en perfusion intraveineuse de 10 minutes, le premier jour de chaque cycle de 21 jours.
Pour les précautions à prendre avant la manipulation ou l'administration d'ALIMTA et pour les instructions concernant la reconstitution et la dilution d'ALIMTA avant administration, voir la rubrique Instructions pour l'utilisation, la manipulation et l'élimination.
Durée de conservation :
Flacon non entamé
3 ans.
Solution reconstituée et solution diluée
Lorsqu'elles sont préparées selon les instructions, les solutions
reconstituées et diluées d'ALIMTA ne contiennent pas de conservateur
antibactérien. La stabilité physique et chimique de la solution
reconstituée et de la solution diluée a été démontrée pendant 24 heures
à température réfrigérée. D'un point de vue microbiologique, le produit
devrait être utilisé immédiatement. S'il n'est pas utilisé
immédiatement, la durée et les conditions de conservation sont sous la
responsabilité de l'utilisateur et ne devraient pas dépasser 24 heures
entre 2°C et 8°C.
Précautions particulières de conservation :
Ce médicament ne nécessite pas de précautions particulières de conservation.
Pour les conditions de conservation du médicament après reconstitution, voir la rubrique Durée de conservation.
Le pemetrexed est physiquement incompatible avec les diluants contenant du calcium, incluant les solutions injectables Ringer et Ringer lactate. En l'absence d'autres études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments.
Reconstituer avec 20 ml de solution de chlorure de sodium à 9 mg/ml (0,9 %) pour préparations injectables, sans conservateur. Les solutions pour perfusion de pemetrexed préparées comme indiqué ci-dessus sont compatibles avec les poches et les tubulures de perfusion intraveineuse en chlorure de polyvinyle (PVC) et polyoléfine.
Le volume approprié de la solution reconstituée de pemetrexed doit être dilué dans 100 ml de solution de chlorure de sodium à 9 mg/ml (0,9 %) pour préparations injectables, sans conservateur.
Les symptômes rapportés en cas de surdosage incluent neutropénie, anémie, thrombopénie, mucite, polyneuropathie sensitive et éruption cutanée. Les complications prévisibles d'un surdosage incluent la dépression médullaire, se manifestant par une neutropénie, une thrombopénie et une anémie. De plus, une infection avec ou sans fièvre, une diarrhée et/ou une mucite peuvent être rapportées. En cas de suspicion de surdosage, la numération formule sanguine des patients doit être surveillée et un traitement symptomatique sera mis en œuvre, selon les cas. L'utilisation d'acide folinique/folinate de calcium dans la prise en charge d'un surdosage de pémétrexed doit être envisagée.
Classe pharmacothérapeutique: Analogues de l'acide folique, Code ATC : L01BA04.
ALIMTA est un agent antinéoplasique antifolate multi-cible qui agit en interrompant des processus métaboliques folate-dépendants essentiels à la réplication cellulaire.
Des études in vitro ont montré que le pémétrexed se comporte comme un anti-folate multi-cible en inhibant la thymidylate synthétase (TS), la dihydrofolate réductase (DHFR) et la glycinamide ribonucléotide formyltransférase (GARFT), qui sont des enzymes folate-dépendantes clés pour la biosynthèse de novo de la thymidine et des nucléotides puriques. Le pémétrexed est transporté dans les cellules à la fois par les systèmes de transport des folates réduits et les protéines membranaires transporteuses de folates. Une fois dans la cellule, le pémétrexed est rapidement et efficacement converti en formes polyglutamates par la folyl-polyglutamate synthétase. Ces formes polyglutamates sont retenues dans les cellules et sont des inhibiteurs encore plus puissants de la TS et de la GARFT. La polyglutamation est un processus temps et concentration-dépendant qui se déroule dans les cellules tumorales et, dans une moindre mesure, dans les tissus normaux. Les métabolites polyglutamatés ont une demi-vie intracellulaire augmentée, prolongeant l'action du produit dans les cellules tumorales.
L'Agence européenne des médicaments a accordé une dérogation à l'obligation de soumettre les résultats d'études réalisées avec ALIMTA dans tous les sous-groupes de la population pédiatrique dans les indications autorisées (voir rubrique Posologie et mode d'administration).
Efficacité clinique
Mésothéliome
L'étude clinique de phase 3 multicentrique, randomisée, en simple aveugle EMPHACIS comparant ALIMTA plus cisplatine versus cisplatine chez les patients atteints de mésothéliome pleural malin n'ayant pas reçu de chimiothérapie antérieure a montré que les patients traités par ALIMTA et cisplatine avaient un avantage cliniquement significatif en terme de survie globale médiane de2,8 mois par rapport aux patients traités par cisplatine seul.
Pendant cette étude, une supplémentation en acide folique à faible dose et en vitamine B12 a été introduite dans le traitement des patients afin d'en réduire la toxicité. L'analyse principale de cette étude a été effectuée sur la population de tous les patients randomisés dans un des bras ayant reçu le traitement correspondant (patients randomisés et traités). Une analyse de sous-groupe a été effectuée chez les patients qui ont reçu une supplémentation en acide folique et en vitamine B12 pendant toute la durée de leur traitement (patients totalement supplémentés). Les résultats d'efficacité de ces analyses sont résumés dans le tableau suivant :
Tableau 5. Résultats d'efficacité d'ALIMTA+cisplatine versus cisplatine dans le mésothéliome pleural malin
| | Patients randomisés et traités | Patients totalement supplémentés | ||
| Paramètre d'efficacité | ALIMTA/ cisplatine (N = 226) | Cisplatine (N = 222) | ALIMTA/ cisplatine (N = 168) | Cisplatine (N = 163) |
| Survie globale médiane (mois) (IC 95 %) | 12,1 (10,0 - 14,4) | 9,3 (7,8 - 10,7) | 13,3 (11,4 - 14,9) | 10,0 (8,4 - 11,9) |
| Test du log-rank (pa) | 0,020 | 0,051 | ||
| Temps médian jusqu'à progression tumorale (mois) (IC 95 %) | 5,7 (4,9 - 6,5) | 3,9 (2,8 - 4,4) | 6,1 (5,3 - 7,0) | 3,9 (2,8 - 4,5) |
| Test du log-rank (pa) | 0,001 | 0,008 | ||
| Temps jusqu'à échec du traitement (mois) (IC 95 %) | 4,5 (3,9 - 4,9) | 2,7 (2,1 - 2,9) | 4,7 (4,3 - 5,6) | 2,7 (2,2 - 3,1) |
| Test du log-rank (pa) | 0,001 | 0,001 | ||
| Taux de réponse globaleb (IC 95 %) | 41,3 % (34,8 - 48,1) | 16,7 % (12,0 - 22,2) | 45,5 % (37,8 - 53,4) | 19,6 % (13,8 - 26,6) |
| Test exact de Fisher (pa) | < 0,001 | < 0,001 | ||
a La valeur de p s'applique à la comparaison entre les bras
b Dans le bras ALIMTA/cisplatine : patients randomisés et traités (N = 225) et patients supplémentés totalement (N = 167)
Une
amélioration statistiquement significative des symptômes cliniquement
importants (douleur et dyspnée) associés au mésothéliome pleural malin
dans le bras ALIMTA/cisplatine (212 patients) comparé au bras
cisplatine seul (218 patients) a été démontrée en utilisant l'échelle
des symptômes du cancer du poumon « Lung Cancer Symptom Scale ».
Des différences statistiquement significatives dans le bilan de la
fonction pulmonaire ont été également observées. La différence entre
les deux bras a été démontrée par l'amélioration de la fonction
pulmonaire dans le bras ALIMTA/cisplatine et la détérioration de
celle-ci au cours du temps dans le bras contrôle.
Les données chez les patients atteints de mésothéliome pleural malin traités par ALIMTA seul sont limitées. ALIMTA a été étudié à la dose de 500 mg/m² en monothérapie chez 64 patients atteints de mésothéliome pleural malin n'ayant jamais reçu de chimiothérapie. Le taux de réponse globale était de 14,1 %.
CBNPC, traitement en seconde ligne
Une étude clinique de phase 3 multicentrique, randomisée, en ouvert comparant ALIMTA versus docétaxel chez des patients atteints de cancer bronchique non à petites cellules (CBNPC), localement avancé ou métastatique, ayant reçu une chimiothérapie antérieure a montré des temps de survie globale médiane de 8,3 mois pour les patients traités par ALIMTA (population en Intention de Traiter n = 283) et de 7,9 mois pour les patients traités par docétaxel (population en Intention de Traiter n = 288). La chimiothérapie antérieure n'incluait pas ALIMTA. Une analyse de l'impact de l'histologie du CBNPC sur l'effet du traitement en termes de survie globale a été réalisée. Les résultats étaient en faveur d'ALIMTA comparé au docétaxel dès lors que l'histologie n'était pas à prédominance épidermoïde (n = 399, 9,3 versus 8,0 mois, HR ajusté = 0,78; 95% IC = 0,61-1,00, p = 0,047) et en faveur du docétaxel dès lors que l'histologie était de type carcinome à cellules épidermoïdes (n = 172, 6,2 versus 7,4 mois, HR ajusté = 1,56; 95% IC = 1,08-2,26, p = 0,018). Aucune différence cliniquement significative n'était observée entre les sous-groupes histologiques concernant le profil de sécurité d'ALIMTA.
Des données cliniques limitées d'une étude à part, de phase 3, contrôlée, suggèrent que les données d'efficacité (survie globale, survie sans progression) de pémétrexed sont similaires entre les patients précédemment prétraités par docétaxel (n = 41) et les patients n'ayant pas reçu de traitement antérieur par docétaxel (n = 540).
Tableau 6. Résultats d'efficacité d'ALIMTA versus docétaxel dans le CBNPC - Population en ITT
| | ALIMTA | Docétaxel | |
| Survie (mois) | (n = 283) | (n = 288) | |
| 8,3 | 7,9 | |
| (7,0 - 9,4) | (6,3 - 9,2) | |
| 0,99 | ||
| (0,82 - 1,20) | ||
| 0,226 | ||
| Survie sans Progression (mois) | (n = 283) | (n = 288) | |
| 2,9 | 2,9 | |
| 0,97 (0,82 - 1,16) | ||
| Temps jusqu'à échec du traitement (mois) | (n = 283) | (n = 288) | |
| 2,3 | 2,1 | |
| 0,84 (0,71 - 0,997) | ||
| Réponse (n : qualifié pour la réponse) | (n = 264) | (n = 274) | |
| 9,1 (5,9 - 13,2) | 8,8 (5,7 - 12,8) | |
| 45,8 | 46,4 | |
Abréviations : IC = intervalle de confiance ; HR : hazard ratio ; ITT : Population en Intention de Traiter ; n = taille population totale.
CBNPC, traitement en première ligne
Une étude clinique de phase 3 multicentrique, randomisée, en ouvert comparant ALIMTA plus cisplatine versus gemcitabine plus cisplatine chez les patients chimio naïfs atteints de CBNPC localement avancé ou métastatique (stade IIIb ou IV) a montré qu'ALIMTA plus cisplatine (population en Intention de Traiter (ITT) n = 862) avait atteint son objectif principal et montrait une efficacité clinique similaire à la gemcitabine plus cisplatine (population en ITT n = 863) en survie globale (hazard ratio ajusté 0,94 ; 95 % IC = 0,84 - 1,05). Tous les patients inclus dans cette étude avaient un Performance Status ECOG de 0 ou 1.
L'analyse primaire d'efficacité était basée sur la population en ITT. Les analyses de sensibilité des principaux critères d'efficacité ont été également évaluées sur la population Qualifiée au Protocole (QP). Les analyses d'efficacité utilisant la population QP sont en accord avec celles utilisant la population en ITT et soutiennent la non-infériorité d'Alimta-cisplatine versus gemcitabine-cisplatine.
La survie sans progression (SSP) et le taux de réponse globale étaient similaires entre les bras de traitement : la médiane de la SSP était de 4,8 mois pour ALIMTA plus cisplatine versus 5,1 mois pour gemcitabine plus cisplatine (hazard ratio ajusté 1,04 ; 95 % IC = 0,94 - 1,15), et le taux de réponse globale était de 30,6 % (95 % IC = 27,3 - 33,9) pour ALIMTA plus cisplatine versus 28,2 % (95 % IC = 25,0 - 31,4) pour gemcitabine plus cisplatine. Les données sur la SSP étaient partiellement confirmées par une revue indépendante (400/1 725 patients étaient sélectionnés au hasard pour cette revue).
L'analyse de l'impact de l'histologie du CBNPC sur la survie globale a démontré des différences cliniquement pertinentes en termes de survie en fonction de l'histologie, voir tableau ci-dessous.
Tableau 7. Résultats d'efficacité d'ALIMTA+cisplatine versus gemcitabine + cisplatine en première ligne de traitement du cancer bronchique non à petite cellule - population en ITT et sous-groupes histologiques.
| Population en ITT et sous- groupes histologiques | Médiane de survie globale en mois (95 % IC) | Hazard ratio (HR) ajusté (95 % IC) | Supériorité valeur p | |||
| ALIMTA + cisplatine | Gemcitabine + cisplatine | |||||
| Population en ITT (N = 1 725) | 10,3 (9,8 - 11,2) | N = 862 | 10,3 (9,6 - 10,9) | N = 863 | 0,94a (0,84 - 1,05) | 0,259 |
| Adénocarcinome (N = 847) | 12,6 (10,7 - 13,6) | N = 436 | 10,9 (10,2 - 11,9) | N = 411 | 0,84 (0,71 - 0,99) | 0,033 |
| Grandes cellules (N = 153) | 10,4 (8,6 - 14,1) | N = 76 | 6,7 (5,5 - 9,0) | N = 77 | 0,67 (0,48 - 0,96) | 0,027 |
| Autre (N = 252) | 8,6 (6,8 - 10,2) | N = 106 | 9,2 (8,1 - 10,6) | N = 146 | 1,08 (0,81 - 1,45) | 0,586 |
| Cellules squameuses (N = 473) | 9,4 (8,4 - 10,2) | N = 244 | 10,8 (9,5 - 12,1) | N = 229 | 1,23 (1,00 - 1,51) | 0,050 |
Abréviations : IC : intervalle de confiance ; ITT : Population en Intention de Traiter ; N = taille population totale.
a statistiquement significatif pour la non-infériorité, un intervalle de confiance entier pour le HR bien en-dessous de 1,17645 fois la marge de non-infériorité (p < 0,001).
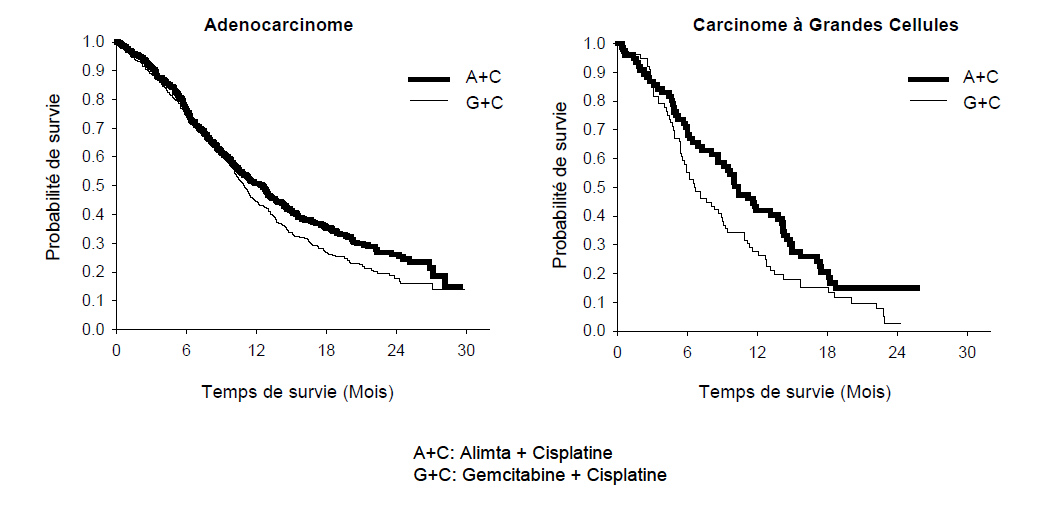
Aucune différence cliniquement significative n'était observée entre les sous-groupes histologiques concernant le profil de sécurité d'ALIMTA plus cisplatine.
Les patients traités avec ALIMTA et cisplatine nécessitaient moins de transfusions (16,4 % versus 28,9 %, p < 0,001), de transfusions de globules rouges (16,1 % versus 27,3 %, p < 0,001) et de transfusions de plaquettes (1,8 % versus 4,5 %, p = 0,002). Les patients nécessitaient également une administration moindre d'érythropoïetine/darbopoïetine (10,4 % versus 18,1 %, p < 0,001), G- CSF/GM-CSF (3,1 % versus 6,1 %, p = 0,004) et de préparations à base de fer (4,3 % versus 7,0 %, p = 0,021).
CBNPC, traitement de maintenance
JMEN
Une étude clinique de phase 3 (JMEN) multicentrique, randomisée, en double-aveugle, contrôlée versus placebo, a comparé l'efficacité et la sécurité du traitement de maintenance par ALIMTA plus meilleurs soins de support (BSC) (n = 441) par rapport au placebo plus meilleurs soins de support (BSC) (n = 222) chez des patients atteints de cancer bronchique non à petites cellules localement avancé (stade IIIB) ou métastatique (stade IV) dont la maladie n'a pas progressé après 4 cycles de traitement en première ligne avec un doublet contenant du cisplatine ou du carboplatine en association avec la gemcitabine, le paclitaxel, ou le docétaxel. Le doublet en traitement de première ligne contenant ALIMTA n'était pas inclus. Tous les patients inclus dans cette étude avaient un Performance Status ECOG de 0 ou 1. Les patients ont reçu le traitement de maintenance jusqu'à progression de la maladie. L'efficacité et la sécurité ont été évaluées dès randomisation après avoir complété le traitement en première ligne (induction). La médiane du nombre de cycles reçus par les patients a été de 5 cycles dans le bras ALIMTA et de 3,5 dans le bras placebo. Un total de 213 patients (48,3 %) a complété ≥ 6 cycles de traitement et un total de 103 patients (23,4 %) a complété ≥ 10 cycles de traitement avec ALIMTA.
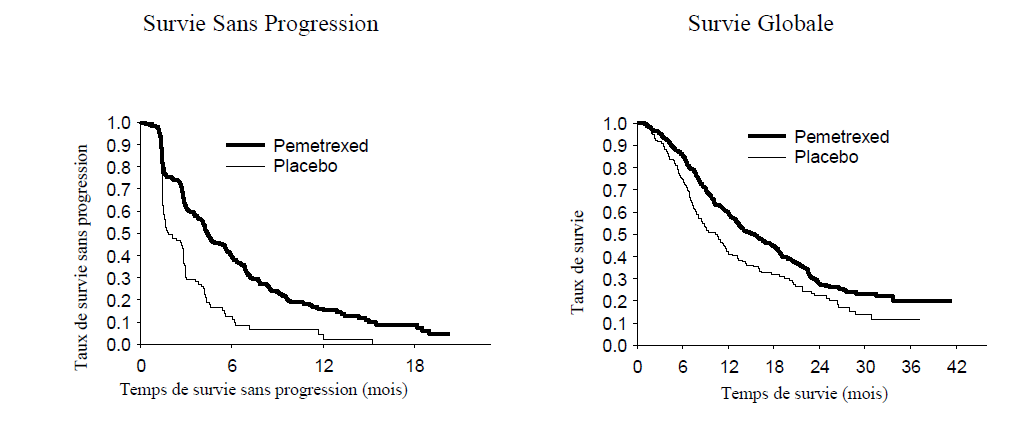
L'étude a atteint son objectif principal et a montré une amélioration statistiquement significative en SSP dans le bras ALIMTA par rapport au bras placebo (n = 581, population revue indépendamment ; médiane de 4,0 mois et 2,0 mois, respectivement) (hazard ratio = 0,60, 95 % IC = 0,49 - 0,73, p < 0,00001). La revue indépendante des scanners des patients a confirmé les conclusions de l'évaluation faite par l'investigateur concernant la SSP. La survie globale médiane pour la population globale (n = 663) était de 13,4 mois pour le bras ALIMTA et 10,6 mois pour le bras placebo, hazard ratio = 0,79 (95 % IC = 0,65 - 0,95 ; p = 0,01192).
En accord avec les autres études cliniques avec ALIMTA, une différence en termes d'efficacité en fonction de l'histologie du CBNPC a été observée dans l'étude JMEN. Pour les patients atteints de CBNPC dès lors que l'histologie n'est pas à prédominance épidermoïde (n = 430, population revue indépendamment) la SSP médiane était de 4,4 mois pour le bras ALIMTA et de 1,8 mois pour le bras placebo, hazard ratio = 0,47 (95 % IC = 0,37 - 0,60, p = 0,00001). La survie globale médiane pour les patients atteints de CBNPC dès lors que l'histologie n'est pas à prédominance épidermoïde (n = 481) était de 15,5 mois pour le bras ALIMTA et 10,3 mois pour le bras placebo, hazard ratio = 0,70 (95 % IC = 0,56-0,88, p = 0,002). En incluant la phase d'induction, la survie globale médiane chez les patients atteints de CBNPC dès lors que l'histologie n'est pas à prédominance épidermoïde était de 18,6 mois pour le bras ALIMTA et 13,6 mois pour le bras placebo, hazard ratio = 0,71 (95 % IC = 0,56-0,88, p = 0,002).
Les résultats sur la SSP et la survie globale chez les patients avec une histologie de type épidermoïde n'ont suggéré aucun avantage pour ALIMTA par rapport au placebo.
Il n'y a pas eu de différences cliniquement pertinentes observées concernant le profil de sécurité d'ALIMTA au sein des sous-groupes histologiques.
JMEN : Graphique de Kaplan Meier sur la survie sans progression (SSP) et la survie globale versus placebo chez les patients atteints de CBNPC dès lors que l'histologie n'est pas à prédominance épidermoïde :
PARAMOUNT
Une étude clinique de phase 3 (PARAMOUNT) multicentrique, randomisée, en double-aveugle, contrôlée versus placebo,
a comparé l'efficacité et la sécurité d'ALIMTA poursuivi en traitement
de maintenance plus meilleurs soins de support (BSC) (n = 359) par
rapport au placebo plus meilleurs soins de support (BSC) (n = 180) chez
des patients atteints de CBNPC localement avancé (stade IIIB) ou
métastatique (stade IV) dès lors que l'histologie n'est pas à
prédominance épidermoïde et dont la maladie n'a pas progressé après 4
cycles de traitement en première ligne avec un doublet ALIMTA associé
au cisplatine. Parmi les 939 patients traités par ALIMTA plus
cisplatine en induction, 539
patients ont été randomisés pour un traitement de maintenance par
pémétrexed ou par placebo. Parmi les patients randomisés, 44,9 %
avaient une réponse complète ou partielle et 51,9 % avaient une maladie
stable suite au traitement ALIMTA plus cisplatine en induction. Les
patients randomisés pour le traitement de maintenance devaient avoir un
Performance Status ECOG de 0 ou 1. La durée médiane entre le début du
traitement ALIMTA plus cisplatine en induction et le début du
traitement de maintenance était de 2,96 mois pour les deux bras de
traitement (bras pémétrexed et bras placebo). Les patients randomisés
ont reçu le traitement de maintenance jusqu'à progression de la maladie.
L'efficacité
et la sécurité ont été évaluées à partir de la randomisation après
avoir complété le traitement en première ligne (induction). Les
patients ont reçu une médiane de 4 cycles de traitement de maintenance
avec ALIMTA et 4 cycles de placebo. Un total de 169 patients (47,1 %)
ont reçu au moins 6 cycles d'ALIMTA en traitement de maintenance, ce
qui représente un total d'au moins 10 cycles d'ALIMTA.
L'étude
a atteint son objectif principal et a montré une amélioration
statistiquement significative de la Survie Sans Progression (SSP) dans
le bras ALIMTA par rapport au bras placebo (n = 472, revue indépendante
de la population ; médianes respectives de 3,9 mois et 2,6 mois)
(hazard ratio = 0,64, IC à 95 % = 0,51 - 0,81, p = 0,0002). La revue
indépendante des scanners des patients a confirmé les conclusions de
l'évaluation faite par l'investigateur concernant la SSP.
Pour
les patients randomisés, la SSP médiane évaluée par l'investigateur,
mesurée depuis le début du traitement par ALIMTA plus cisplatine en
induction, était de 6,9 mois pour le bras ALIMTA et 5,6 mois pour le
bras placebo (hazard ratio = 0,59, IC à 95 % = 0,47 - 0,74).
Suite à un traitement d'induction avec ALIMTA plus cisplatine (4 cycles), le traitement par ALIMTA était statistiquement supérieur au placebo pour la Survie Globale (SG) (médiane de 13,9 mois versus 11,0 mois, hazard ratio = 0,78, 95% IC=0,64-0,96, p=0,0195). Au moment de l'analyse finale de la survie, 28,7% des patients étaient en vie ou perdus de vue dans le bras ALIMTA versus 21,7% dans le bras placebo. L'effet relatif du traitement par ALIMTA était constant au sein des sous-groupes (incluant le stade de la maladie, la réponse à l'induction, le Performance Status ECOG, le statut de fumeur, le sexe, l'histologie et l'âge) et similaire à celui observé dans les analyses de SG et SSP non ajustées. Les taux de survie à 1 an et à 2 ans des patients sous ALIMTA étaient de 58% et 32% respectivement, comparés à 45% et 21% pour les patients sous placebo. Depuis le début du traitement d'induction de première ligne avec ALIMTA plus cisplatine, la SG médiane des patients était de 16,9 mois pour le bras ALIMTA et de 14,0 mois pour le bras placebo (hazard ratio=0,78, 95% IC=0,64- 0,96). Le pourcentage des patients ayant reçu un traitement après l'étude était de 64,3% pour ALIMTA et 71,7% pour le placebo.
PARAMOUNT : Graphique de Kaplan Meier sur la survie sans progression (SSP) et la survie globale (SG) pour ALIMTA poursuivi en maintenance versus placebo chez les patients atteints de CBNPC dès lors que l'histologie n'est pas à prédominance épidermoïde (mesurée depuis la randomisation) :
Survie Sans Progression Survie Globale
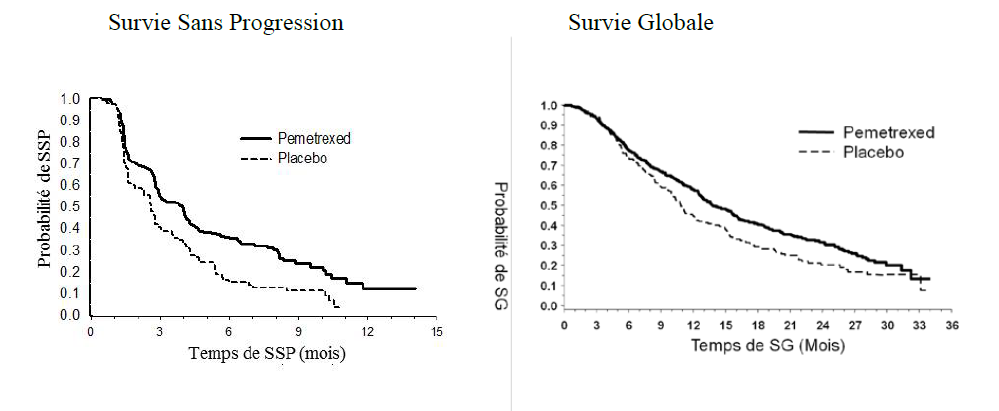
Les profils de sécurité d'ALIMTA en maintenance dans les 2 études JMEN et PARAMOUNT étaient similaires.
Les
paramètres pharmacocinétiques du pémétrexed en monothérapie ont été
évalués chez 426 patients cancéreux présentant différentes tumeurs
solides recevant des doses de 0,2 à 838 mg/m2, en perfusion de 10 minutes. Le pémétrexed a un volume de distribution à l'équilibre de 9 L/m². Des études in vitro indiquent
que le pémétrexed est lié à environ 81 % aux protéines plasmatiques.
Cette liaison n'est pas notablement modifiée par une insuffisance
rénale, de quel que degré qu'elle soit. Le métabolisme hépatique du
pémétrexed est limité. Le pémétrexed est principalement éliminé dans
les urines, 70 % à 90 % de la dose étant retrouvée inchangée dans les
urines des premières 24 heures suivant l'administration. Les études in vitro indiquent
que le pémétrexed est activement sécrété par OAT3 (transporteur
d'anions organiques 3). La clairance systémique totale est de 91,8
mL/min et la demi-vie d'élimination est de 3,5 heures chez les patients
ayant une fonction rénale normale (clairance de la créatinine de 90
mL/min). La variabilité inter-individuelle de la clairance est modérée
(19,3 %).
L'exposition systémique totale (aire sous la courbe - AUC) et la concentration maximale (Cmax) augmentent
proportionnellement à la dose. Les caractéristiques pharmacocinétiques
du pémétrexed sont constantes d'un cycle à l'autre.
Les paramètres pharmacocinétiques du pémétrexed ne sont pas influencés par l'administration concomitante de cisplatine. La supplémentation en acide folique par voie orale et en vitamine B12 par voie intramusculaire n'affecte pas la pharmacocinétique du pémétrexed.
Les effets sur l'aptitude à conduire des véhicules et à utiliser des machines n'ont pas été étudiés. Toutefois, il a été rapporté que le pémétrexed pouvait causer une fatigue. Si cet effet se produit, les patients doivent éviter de conduire des véhicules et d'utiliser des machines.
L'administration de pémétrexed à des souris gravides s'est traduite par une diminution de la viabilité des fœtus, une diminution du poids des fœtus, une ossification incomplète du squelette et des fentes palatines.
L'administration de pémétrexed chez des souris mâles a eu des effets toxiques sur la reproduction, caractérisés par une réduction de la fertilité et par une atrophie testiculaire. Dans une étude conduite chez le chien beagle avec injection de bolus intraveineux pendant 9 mois, des effets sur les testicules (dégénérescence/nécrose de l'épithelium séminifère) ont été observés. Cela suggère que le pémétrexed peut altérer la fertilité masculine. La fertilité féminine n'a pas été étudiée.
Le pémétrexed n'a pas montré de potentiel mutagène que ce soit dans le test d'induction d'aberrations chromosomiques in vitro sur cellules d'ovaire de hamster chinois (CHO) ou dans le test d'Ames. Le pémétrexed s'est montré clastogène dans le test in vivo sur micronoyaux de souris.
Il n'a pas été conduit d'étude sur le potentiel carcinogène du pémétrexed.
Utiliser des techniques aseptiques pour la reconstitution et la dilution ultérieure de la solution de pémétrexed pour administration par perfusion intraveineuse.
Calculer la dose et le nombre de flacons d'ALIMTA nécessaires. Chaque flacon contient un excès de pémétrexed pour faciliter l'administration de la quantité prescrite.
ALIMTA 100 mg
Reconstituer le flacon de 100 mg avec 4,2 mL de solution de chlorure de sodium à 9 mg/mL (0,9 %) pour préparations injectables, sans conservateur, ce qui donne une solution contenant environ 25 mg/mL de pémétrexed.
ALIMTA 500 mg
Reconstituer le flacon de 500 mg avec 20 mL de solution de chlorure de sodium à 9 mg/mL (0,9 %) pour préparations injectables, sans conservateur, ce qui donne une solution contenant environ 25 mg/mL de pémétrexed.
Agiter délicatement chaque flacon jusqu'à ce que la poudre soit complètement dissoute. La solution ainsi obtenue est claire et sa couleur varie de l'incolore au jaune ou jaune verdâtre sans conséquence sur la qualité du produit. Le pH de la solution reconstituée varie de 6,6 à 7,8. Une dilution ultérieure est nécessaire.
Le volume approprié de la solution reconstituée de pémétrexed doit être à nouveau dilué pour atteindre 100 mL, avec une solution de chlorure de sodium à 9 mg/mL (0,9 %) pour préparations injectables, sans conservateur, et administré en perfusion intraveineuse de10 minutes.
Les solutions pour perfusion de pémétrexed préparées comme indiqué ci-dessus sont compatibles avec les poches et les tubulures de perfusion intraveineuse en chlorure de polyvinyle (PVC) et polyoléfine.
Les médicaments pour usage parentéral doivent faire l'objet d'une inspection visuelle avant administration, pour détecter la présence éventuelle de particules ou d'une modification de la couleur. Si des particules sont présentes, ne pas administrer.
Les solutions de pémétrexed sont à usage unique seulement. Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Préparation et précautions d'administration
Comme pour tout agent anticancéreux potentiellement toxique, des précautions doivent être prises lors de la manipulation et de la préparation des solutions pour perfusion de pémétrexed. L'utilisation de gants est recommandée. En cas de contact de la solution de pémétrexed avec la peau, laver la peau immédiatement et abondamment avec de l'eau et du savon. En cas de contact de la solution de pémétrexed avec les muqueuses, rincer abondamment avec de l'eau. Le pémétrexed n'est pas un agent vésicant. Il n'existe pas d'antidote spécifique en cas d'extravasation de pémétrexed. Quelques cas d'extravasation de pémétrexed ont été rapportés et ont été considérés comme non graves par les investigateurs. Les extravasations devraient être prises en charge selon les pratiques standard locales appliquées aux autres agents non-vésicants.
liste I
médicament nécessitant une surveillance particulière pendant le traitement
prescription hospitalière
prescription réservée aux spécialistes et services CANCEROLOGIE
prescription réservée aux spécialistes et services HEMATOLOGIE
prescription réservée aux spécialistes et services ONCOLOGIE MEDICALE
médicament nécessitant une surveillance particulière pendant le traitement
prescription hospitalière
prescription réservée aux spécialistes et services CANCEROLOGIE
prescription réservée aux spécialistes et services HEMATOLOGIE
prescription réservée aux spécialistes et services ONCOLOGIE MEDICALE
Poudre pour solution à diluer pour perfusion.
Poudre lyophilisée de couleur blanche à jaune pâle ou jaune verdâtre.
Flacon de verre de type I avec un bouchon en caoutchouc, contenant 500 mg de pemetrexed.
Boîte de 1 flacon.
ALIMTA 500 mg poudre pour solution à diluer pour perfusion
Chaque flacon contient 500 mg de pémétrexed (sous forme de pémétrexed disodique).
Excipient à effet notoire :
Chaque flacon contient approximativement 54 mg de sodium.
Après reconstitution (voir rubrique Instructions pour l'utilisation, la manipulation et l'élimination), chaque flacon contient 25 mg/mL de pémétrexed. Pour la liste complète des excipients, voir rubrique Liste des excipients.
Mannitol
Acide chlorhydrique Hydroxyde de sodium